洁净度级别确认 | 分级双粒径0.5/5.0微米技术逻辑解析,尘埃粒子计数器为何大流量选型的成了重大底层误区

洁净度级别确认 | 分级双粒径0.5/5.0微米技术逻辑解析,尘埃粒子计数器为何大流量选型的成了重大底层误区
· 导语 ·
国内《药品生产质量管理规范附录一(无菌药品)》2026 修订版目前处于内部征求意见阶段,尚未正式颁布落地执行,但本次修订第七章「洁净区确认与监测」对标 EU GMP Annex 1(2023 版)、ISO 14644-1 分级体系,明确提出 ABCD 全级别洁净区在静态分级确认、动态持续监控、周期性再确认环节,同步采集≥0.5μm、≥5.0μm 两类悬浮粒子浓度数据的监管导向。
当前大量制药企业、第三方验证机构仅聚焦采样流量参数选型,忽略粒径检测分辨率与计数效率的硬性约束,极易在前期预合规整改、药监阶段性检查中形成系统性偏差。本文从修订条款技术要求、采样量计算逻辑、仪器选型底层误区、质量风险维度逐层拆解双粒径监测的合规内涵。
· 一、新版附录一征求意见稿核心技术导向:双粒径并行监测成为分级验证刚性框架 ·
|
2026 版 GMP 无菌附录一 第七十八条 洁净区级别确认是洁净区确认的一部分。洁净区的级别确认时应当分别测量 ≥0.5μm 和 25um 的颗粒的总数。测试需要在静态和动态操作环境中进行。级别确认活动应当避免对工艺或产品质量造成任何影响。初始级别确认建议在模拟操作期间进行,再次进行级别确认时建议在模拟操作期间或无菌工艺模拟试验期间进行。各洁净级别悬浮粒子的标准规定如下表:
注: (1) 如有 CCS 或历史趋势数据依据,可考虑将 25um 颗粒总数作为分级的参考标准。 (2) D 级的动态限度不作统一规定。企业应当根据风险评估和日常监测的数据确定动态限度。 |
|||||||||||||||||||||||||||||||||||

现行有效 GMP 附录一已对 A/B 高风险洁净区执行双粒径监测要求,2026 修订征求意见稿进一步将该要求拓展至 C/D 辅助洁净区,取消分级监测的粒径豁免条款:洁净室洁净度判定需同时满足≥0.5μm、≥5.0μm 两项浓度限值,双指标同时合规方可完成级别确权与放行使用。两类粒径承担差异化污染判定职能,共同构成无菌环境风险管控闭环:
1、≥0.5μm 亚微米级颗粒:核心用于评定围护结构、高效过滤系统的基础净化效能,是洁净室基础静态分级的核心指标,反映空调净化系统整体密封与过滤性能。
2、≥5.0μm 大颗粒:主要来源于人员作业扬尘、物料碎屑、设备摩擦脱落、气流紊流裹挟杂质等动态人为风险,是活性微生物的实时指标粒径:动态生产场景下染菌、异物污染的核心溯源指标。
本次修订拓宽双粒径适用范围,本质是监管逻辑从「静态硬件达标」升级为「动态生产风险量化管控」,依托双维度粒子数据搭建 CCS 污染控制策略的数据底座,对监测仪器的数据溯源性、测量精准度提出标准化约束。
· 二、0.5/5.0微米双粒径体系下采样体积重构:大流量采样成为统计有效性的前置条件 ·
依照 ISO 14644-1 分级公式V_s=(20/洁净度级别)×1000,洁净区单点最小采样体积由限值更严苛、粒子本底浓度更低的粒径决定,需单测点至少采集 20 个有效粒子以满足 95% 置信区间统计学判定要求。
制药 A 级洁净区≥5.0μm 粒子浓度上限仅 20 粒 /m³,经核算单点最低采样体积需达到 1000L;传统 50L/min 流量设备单点采样时长超 20 分钟,多点位批量分级验证效率严重不足。行业由此形成共识:高等级洁净区分级验证必须选用大流量粒子计数器,以此压缩采样时长、满足批量点位验证需求。
· 三、行业选型共性偏差:唯流量参数导向,偏离 ISO 21501-4 计数效率校准规范 ·
现阶段业内普遍陷入流量单一维度选型误区:优先选用 100L/min 大流量机型,以高流量缩短采样时长,但忽略仪器最小可测粒径、计数效率双通道约束,最终出现采样流程合规、测量数据无效的结构性问题。
依据 ISO 21501-4:2018 光散射粒子计数器校准规范(新版GMP指向的权威粒子计数器校准标准),仪器需满足双通道计数效率限值:
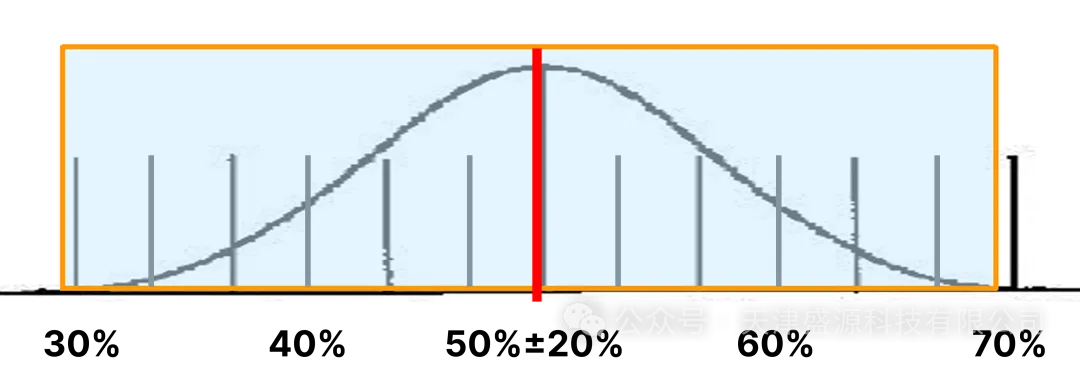
近最小灵敏度粒径(第一粒径):计数效率 50%,允许相对偏差 ±20%;
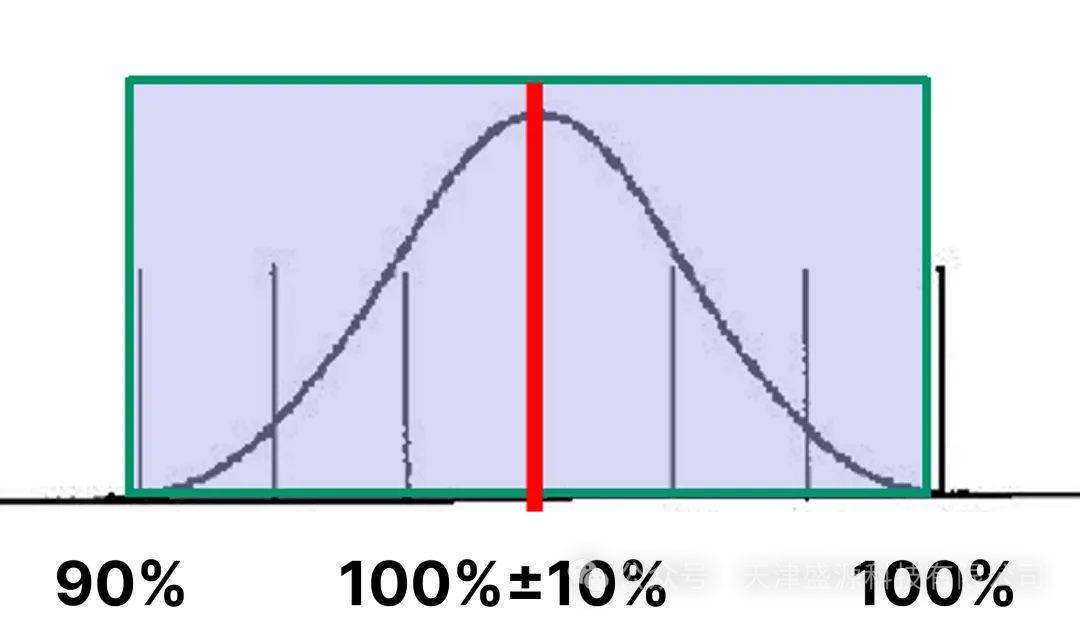
1.5~2 倍最小灵敏度粒径(第二工作粒径):计数效率100%,允许相对偏差仅 ±10%。
制药行业日常验证以 0.5μm、5.0μm 为核心工作通道,其中 0.5μm 属于第二粒径,必须依托≤0.3μm 的最小可测粒径作为光学分辨基底,方可将计数效率稳定锁定在 90%~110% 合规区间。市面主流 100L/min 大流量设备最小可测粒径仅为 0.5μm,无 0.3μm 光学分辨能力,0.5μm 通道无法满足 100% 计数效率的校准要求,粒径分辨率不足会引发粒子漏检、浓度数值漂移,整套分级监测数据不具备审计采信效力。
· 四、测量精度失准衍生的系统性质量与合规风险 ·
仪器最小可测粒径局限带来的误差,并非单纯数值偏差问题,会逐层传导至验证、生产、审计全链条:
1、洁净等级误判风险:偏低的计数效率会低估 0.5μm 粒子浓度,将实际超标的洁净区判定为合规等级,不合格净化环境投入无菌生产,极易引发药液微粒超标、无菌灌装染菌、批次报废等重大质量事故。
2、过滤器失效无法溯源:0.5μm 通道数据失真,无法精准判定高效过滤器密封渗漏、滤材破损等问题,过滤系统隐患长期隐匿。
3、审计不符合项风险:药监核查、第三方 CMA 验证审计会核验仪器校准资质与硬件参数,若设备不满足 ISO 21501-4 粒径分辨要求,会直接判定环境监测体系存在重大缺陷,要求停产重验证、整改质量体系,产生高额时间与运营成本。
· 仪器选型合规基准总结 ·
结合附录一修订征求意见稿、ISO 14644-1、ISO 21501-4 三重标准约束,合规粒子计数器必须同时满足两大硬性维度:
1. 采样流量维度:流量匹配 5.0μm 粒径大体积采样需求,保障单点采样统计学有效性与现场作业效率;
2. 光学精度维度:最小可测粒径≤0.3μm,锚定 0.5μm 通道 100% 计数效率,偏差控制在 ±10% 以内。
流量与精度二者不可偏废,片面追求大流量而牺牲光学分辨下限,是当前药企预合规整改最普遍的技术盲区;只有双维度指标同时达标,监测数据才可用于分级确认、趋势分析、审计溯源,真正以量化数据支撑污染控制策略落地,筑牢无菌生产环境底线。
免责声明
· 本公众号部分观点、内容非原创,仅作信息转载与传播之用,所载观点不代表本公众号立场。
· 平台发布的技术知识、行业资讯,仅限个人学习、知识留存使用,禁止任何商业转载与商用行为。
· 若有版权纠纷或其他问题,请及时与我们联系,我们将尽快核实并妥善处理。